I retrieved expression data of muscarinic receptor gene CHRM1 - 5 in all neuron cells from the WHB-10Xv3 dataset. Attached figure 1 plotted gene expression in CPM and percent of detection, where CHRM4 expression was barely detected.
CHRM4 is known to be expressed at relatively abundant level comparing to other CHRM genes in selected brain regions. See attached figure 2 and 3 for gene expression in bulk brain tissues (GTEx data) and protein expression (Human Protein Atlas).
What may be the reason for the seeming lack of CHRM4 expression detection in the WHB-10Xv3 dataset. Does CHRM4 truly have lower expression than the other CHRM genes in neurons of the measured brain regions, or some factors contributed to its under estimation? There may be factors such as 10X Genomic platform, experiment QC, brain sample collection, CHRM4 sequence, … Can you please help assess it?
The short answer is that I don’t know, however you are not the first person to notice this happening in one of our data sets for a gene (e.g., this post: Known protein not detected by RNA explorer).
I think any of your suggestions are reasonable. In some cases the differences are technical, although it seems unlikely here since at least three human and one mouse data sets show similarly low expression of CHRM4 across most cells–so also not likely a cross-species difference. Another possibility (which I don’t think is likely given how transcripts are selected) is that the reads for CHRM4 might not be properly aligned with the location of the gene annotation in the genome. We can see in both MTG and M1 that reads pile up in both exonic and intronic locations.
What we can safely say is that CHRM4 does not have high expression in very many nuclei collected from human brains across a variety of studies. Finally, while it is possible that CHRM4 is expressed more highly in single cells rather than single nuclei, this does not appear to be the case in mouse (Single-nucleus and single-cell transcriptomes compared in matched cortical cell types).
I’m not sure what possibilities this leaves (maybe previous studies were mistaken?), but hopefully this post at least provides some directions to pursue and rules out some options.
Thank you very much for your response, and apologies for my delayed follow-up!
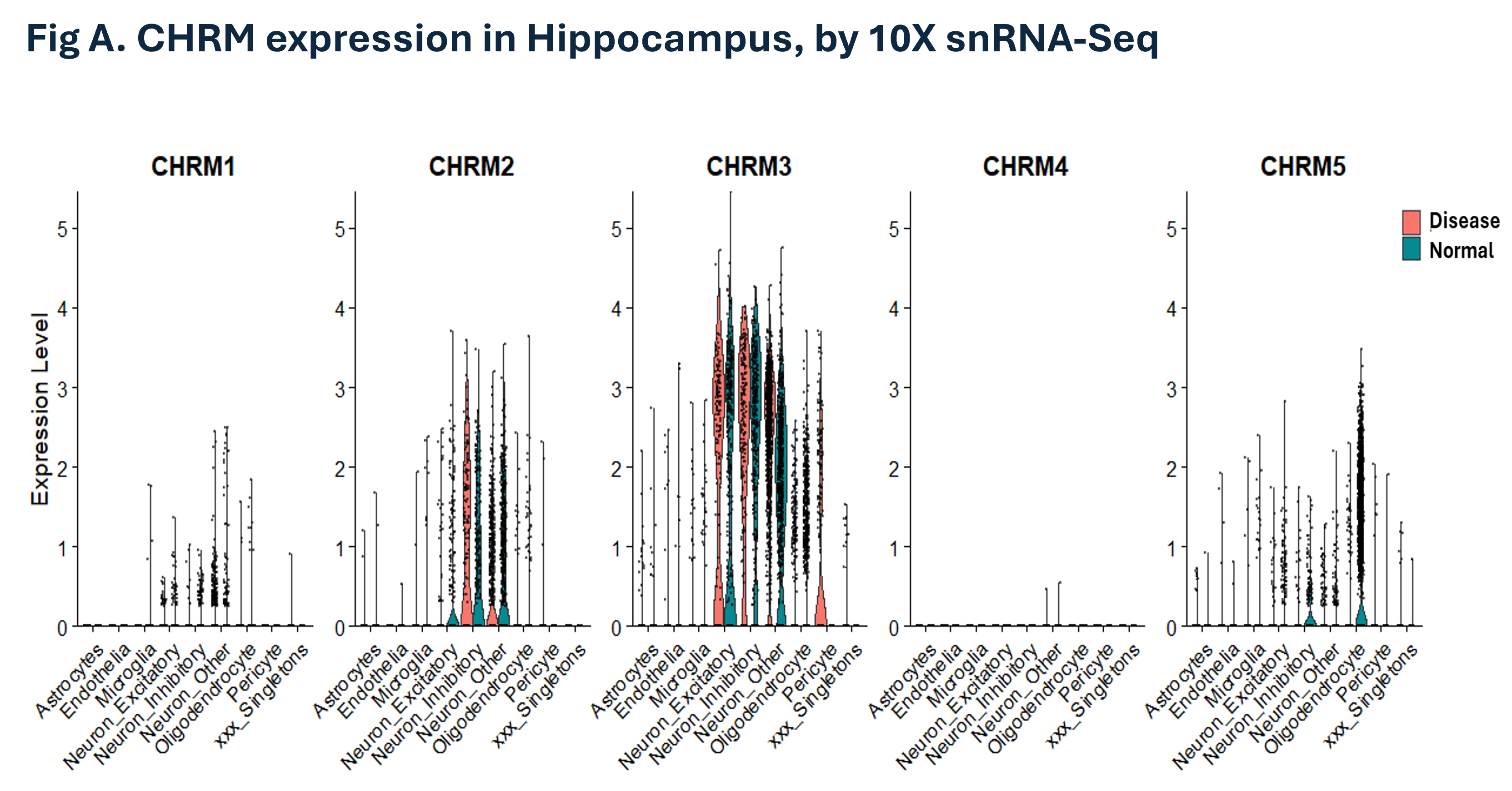
The WHB-10Xv3 dataset has high data quality. We were able to replicate the CHRM detection profile in an independent snRNA-seq dataset (Fig. A).
I was wondering whether differences in post-transcriptional processing and nuclear-to-cytoplasmic transporting among transcripts could affect the measurements. Figure 5H from your reference highlights nucleus-versus-soma proportion differences across example transcripts. We retrieved the data sets and compared CHRM expression between single-cell and single-nucleus datasets, and indeed observed higher CHRM4 detection in cells than in nuclei (Fig. B).
These preliminary results suggest that transcript processing may influence nuclear versus cytoplasmic proportions. Since snRNA-seq is widely used for brain samples by Allen and many others, would Allen be interested in systematically comparing nuclear and cellular RNA-seq datasets and developing a conversion/normalization factor for each transcript to more accurately infer cellular transcript abundance from snRNA-seq data?
Given your analysis, it seems quite plausible that it’s a cell vs. nucleus difference, as you suggest. Several years ago we did compare cellular and nuclear transcript abundance from matched cells in mouse visual cortex (here, and linked above), but technologies have improved and data sets have massively expanded since then, so it would be quite reasonable to do it again.
Regarding this:
I think it would be an excellent resource for the community and I would strongly advocate its creation. To the best of my knowledge, such a resource does not yet exist, nor is it planned by Allen Institute scientists. If you (or others on the Community Forum) create such a resource, I’d encourage you to link it here.
Great to hear that you also see the value of such a resource. I also agree that we’ll need fresh data for broader coverage and to keep pace with technological advances. Unfortunately, we don’t have the capacity to generate new datasets.
Allen Institute not only has the capacity, but more importantly the credibility in the community to lead this effort. I’d be happy to support on data analysis and other downstream work. Please let me know what you and others at Allen think, and whether it would be helpful for us in the community to petition Allen to advocate for it.
I don’t think data generation would be necessary to create this resource. For example, the Allen Institute now shares matched data for a subset of brain regions using both single cell and single nucleus transcriptomics, presented in Yao et al 2023 and Langlieb et al 2023 studies, respectively. Similarly matched data sets in human (albeit likely only in selected neuronal cell types) could potentially also be gathered, although I am not aware of a place where this has already been done.
While there is plenty more data that will be coming from the Allen Institute moving forward, we are not taking specific external requests at this time.
I was thinking more along the lines of single-cell and single-nucleus data from the same sample sets and by the same lab, to minimize potential confounders. Thanks for your reference pointing out that Allen has already generated such a dataset. I’ll look into it.