Hello,
I created a query data set using SEA-AD snRNAseq MTG data from the raw fastqs at sage Synape (with clinical consensus diagnosis of Alzheimers disease and Control). I ran cell ranger (with introns) and perfomed QC. The ref I am using the MTG final_nuclei ref provided in the AWS registry by Gabitto et al.
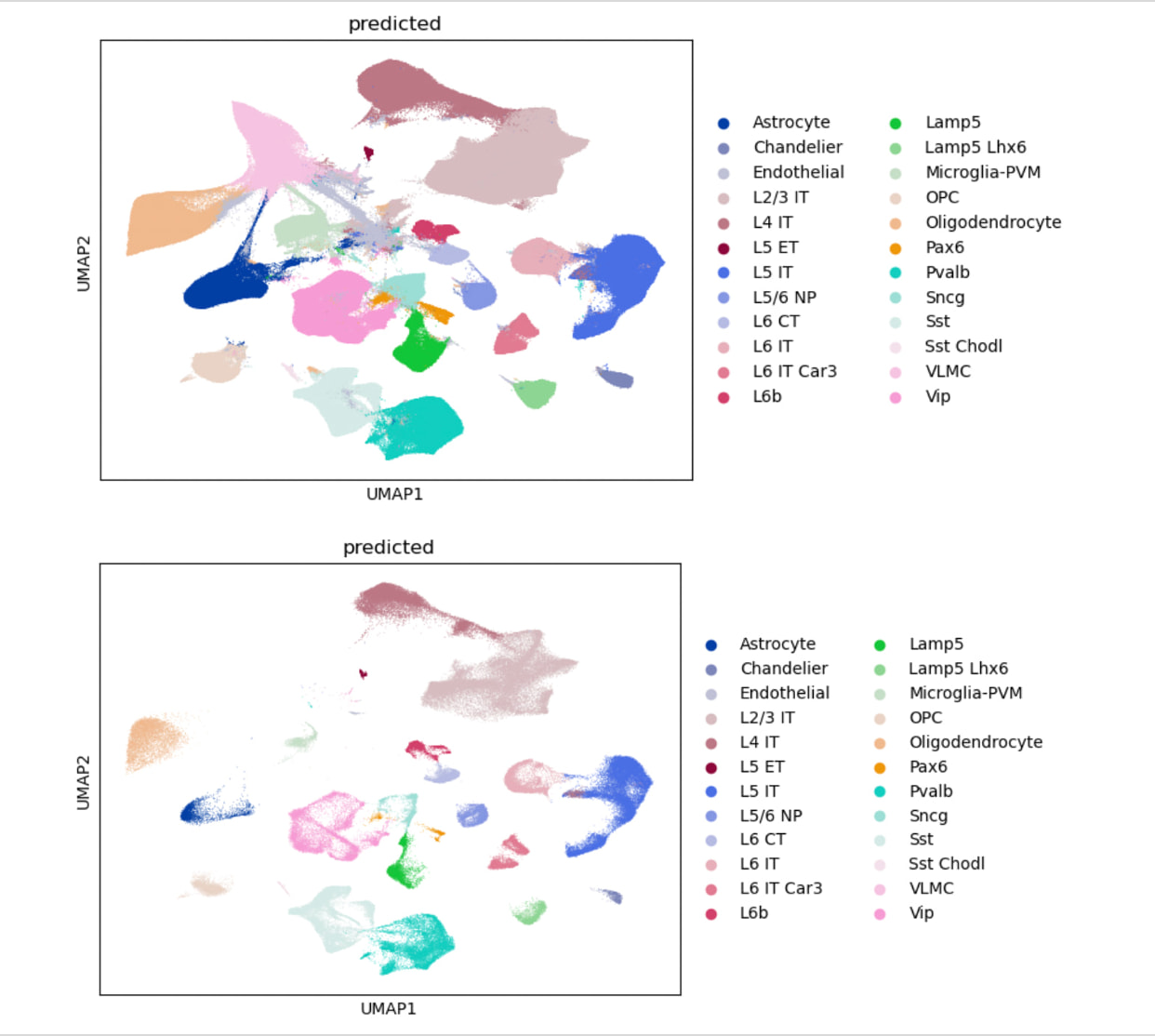
I have large portion of the query that wont match the ref.
(TOP )QUERY+REF. (BOTTOM) JUST REF (same latent space)
I’m using this for my scvi:
scvi.model.SCVI.setup_anndata(adata, batch_key=“libraryBatch”, layer=“counts”,
categorical_covariate_keys=[“individualID”, “sex”],
continuous_covariate_keys=[“age_numeric”])
vae=scvi.model.SCVI(adata, n_latent=30)
And this for my scANVI
lvae=scvi.model.SCANVI.from_scvi_model(vae,adata=adata,
unlabeled_category=“Unknown”,
labels_key=“subclass_label”)
lvae.train(max_epochs=100, early_stopping=True, n_samples_per_label=100)
I have added/substacted various other categorical covariates and tried to fix this issue but the result is the same. The cluster at the top left of the query+ref UMAP also gets assigned a different cell type with subtle changes in the categorical covariates as well.
Any help/suggestions would be appreciated.
1 Like
Hi @ashaypatel,
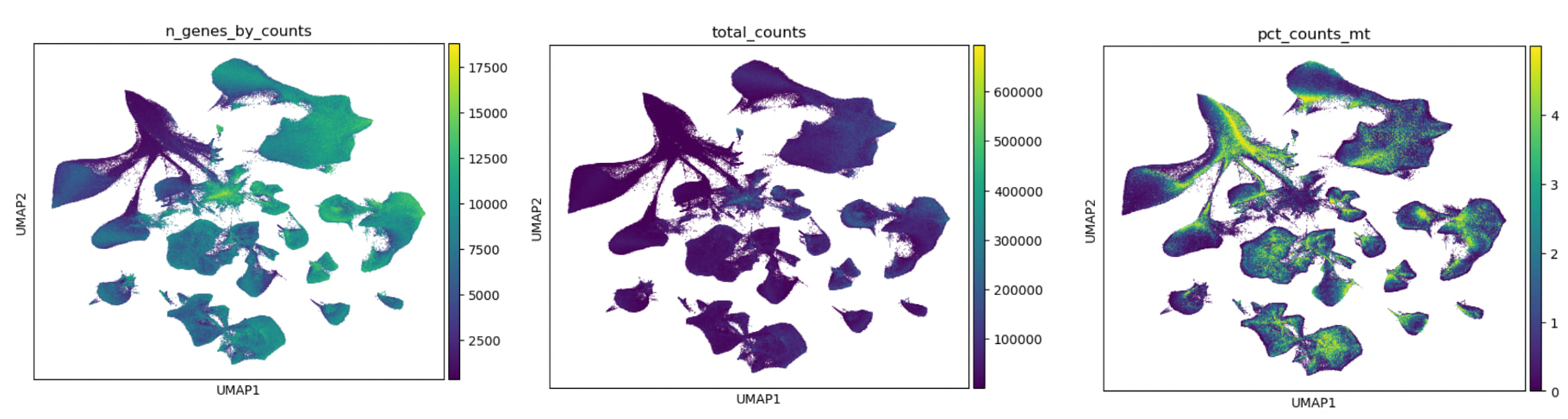
That looks suspiciously like low quality nuclei. Have you plotted the number of genes detected, UMIs, or fraction of mitochondrial UMIs on your representation?
Best, Kyle
1 Like
Hello @kyle.travaglini , thanks for your response
I used mitochondrial filter of <5%, ribosomal filter of <5% and hemoglobin filter of <1%. For filter cells i kept genes with min_genes>250 (perhaps too leniant?) and sc.pp.filter_genes(adata, min_cells=25).
What do you suggest?
1 Like
Here are the plots. I was wondering if I should at pct_counts_mt as a categorical covariate in scVI? Additionally n_genes_by_counts does show lower gene detection in the problematic quadrant of the UMAP however, it is only “removed“ when I do data = adata[adata.obs[‘n_genes_by_counts’] >= 2000, :] in the same latent space, which is really stringent.
1 Like
~2000 can be quite low depending on the cell type, most neurons have a ton of RNA. The other clue these are low quality is the high mitochondrial fraction. We also sequence our samples more deeply than most and use higher gene cutoffs (>1500) for all other types except Microglia (>1000). For reference: SEA-AD_2024/Single nucleus omics/01_Mapping and Quality Control/RNAseq/iterative_scANVI.py at main · AllenInstitute/SEA-AD_2024 · GitHub & SEA-AD_2024/Single nucleus omics/01_Mapping and Quality Control/RNAseq/00_MTG mapping and QC.ipynb at main · AllenInstitute/SEA-AD_2024 · GitHub -Kyle
Thanks you for getting back to me. Much appreciated